LAB MEMBERS
박사/석박사



이기주 2017~ 김민석 2018~ 김태건 2018~
석사



박세리 2022~ 김경임 2022~ 박지원 2024~
Alumni


 .
. 

김명호 석사2021 박성민 석사2021 전우석 석사2021 박두호 석사2021 오원석 석사2023
2021 이윤정, 정일용
2020 박용준, 송승민
2019 박세진, 최주헌
2018 황선영, 오창욱
MARS: leveraging allelic heterogeneity to increase power of asswociation testing
In standard genome-wide association studies (GWAS), the standard association test is underpowered to detect associations between loci with multiple causal variants with small effect sizes. We propose a statistical method, Model-based Association test Reflecting causal Status (MARS), that finds associations between variants in risk loci and a phenotype, considering the causal status of variants, only requiring the existing summary statistics to detect associated risk loci. Utilizing extensive simulated data and real data, we show that MARS increases the power of detecting true associated risk loci compared to previous approaches that consider multiple variants, while controlling the type I error.
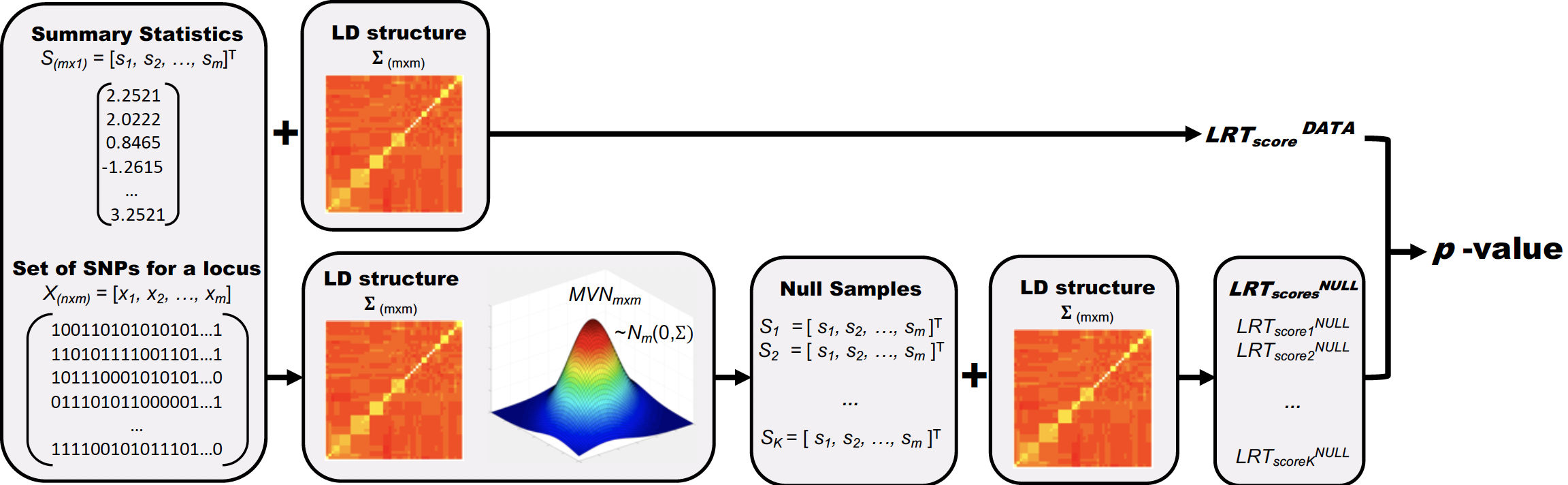
Overview of MARS. Here, we assume that we are testing an association between a locus of m variants and a trait. The leftmost panel shows the input of MARS; m number of summary statistics for the locus and an n×m matrix that contains genotypes of m SNPs for n samples. The next two panels on the bottom show the re-sampling process in which we sample the null statistics K times from an MVN distribution with a variance-covariance matrix of Σ that contains LD of the genotypes X. The rightmost panel shows the process by which we estimate LRTstats for the null panel from which we can compute a p-value for the data.
The full citation to our paper is: Hormozdiari, F., Jung, J., Eskin, E., Jong Wha J. Joo. MARS: leveraging allelic heterogeneity to increase power of association testing. Genome Biol 22, 128 (2021). https://doi.org/10.1186/s13059-021-02353-8
Multiple testing correction in linear mixed models
Background
Multiple hypothesis testing is a major issue in genome-wide association studies (GWAS), which often analyze millions of markers. The permutation test is considered to be the gold standard in multiple testing correction as it accurately takes into account the correlation structure of the genome. Recently, the linear mixed model (LMM) has become the standard practice in GWAS, addressing issues of population structure and insufficient power. However, none of the current multiple testing approaches are applicable to LMM.
Results
We were able to estimate per-marker thresholds as accurately as the gold standard approach in real and simulated datasets, while reducing the time required from months to hours. We applied our approach to mouse, yeast, and human datasets to demonstrate the accuracy and efficiency of our approach.
Conclusions
We provide an efficient and accurate multiple testing correction approach for linear mixed models. We further provide an intuition about the relationships between per-marker threshold, genetic relatedness, and heritability, based on our observations in real data.
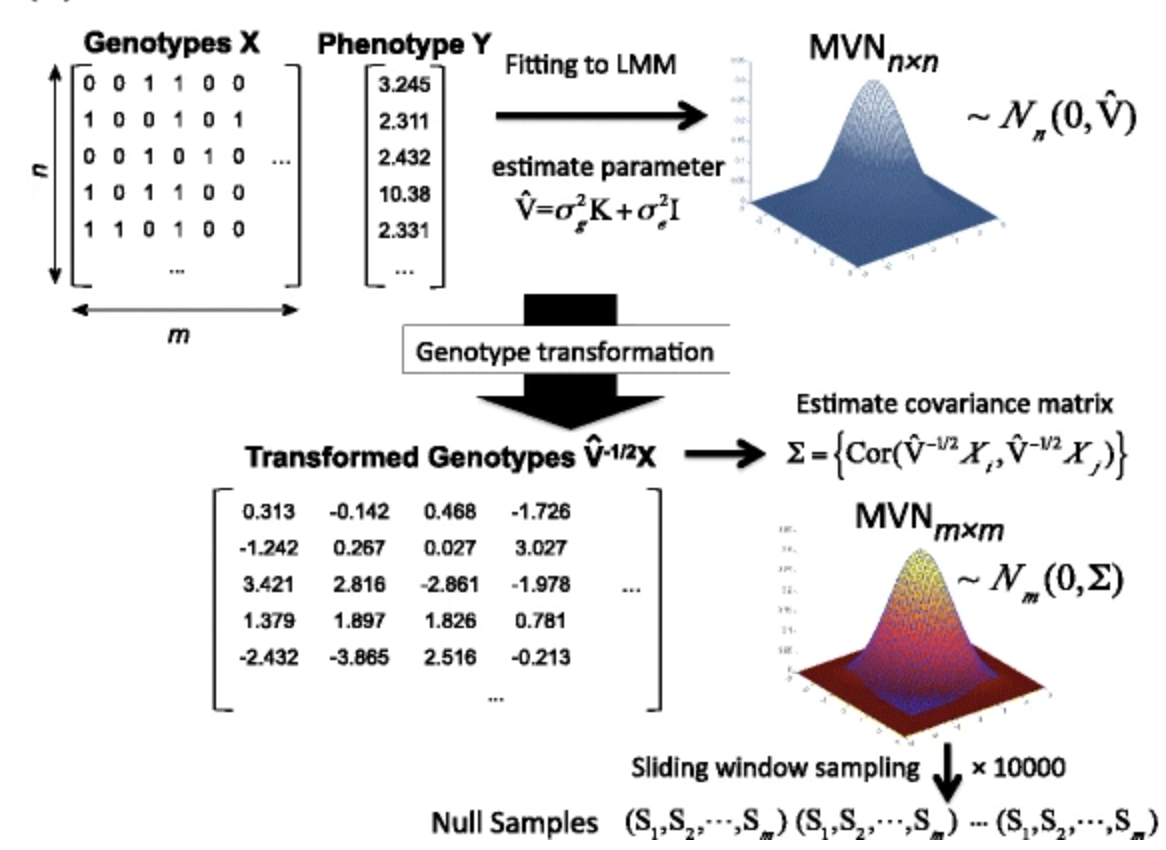
Overview of the resampling procedure of MultiTrans, 10000 sampling applied
The full citation to our paper is: Joo, Jong Wha; Hormozdiari, Farhad; Han, Buhm; Eskin, Eleazar, Multiple testing correction in linear mixed models, Genome Biol, 17 (1), pp. 62, 2016, ISSN: 1474-760X.